


由伤寒和副伤寒沙门菌所致的伤寒热能引起暴发流行,尤其在缺乏安全供水和食物的地区,可形成严重的公共卫生问题。另外由非伤寒沙门菌导致的侵袭性感染,在诸如非洲一些国家,也造成一定的疾病负担。伤寒热在非洲多个地区报道发病率高,但因为缺少准确的实验室病原学鉴定依据,而常使用的肥达反应虽然简便,因敏感度和特异性都偏低,所以基于肥达反应推算的伤寒热疾病负担是不准确的。
塞拉利昂属于西非国家,每年报道有大量的伤寒热病例,但这些病例均为基于肥达反应或临床症状诊断,没有基于病原学的调查研究。为了了解塞拉利昂的伤寒热疾病负担以及肥达反应的诊断效能,由中国疾控中心和塞拉利昂卫生部共同组织、中塞友好固定生物安全实验室承担,于2019年在塞拉利昂首府弗里敦市开展了发热病人伤寒加强监测项目,设立了7家哨点医院,选择发热两天及以上的病人,采集血液样品进行细菌血培养鉴定。研究中设立了严格的检测程序,包括分别于培养的3天和7天取培养上清进行沙门菌选择性培养基接种,并且在培养的7天和14天抽取培养瓶的培养液进行荧光PCR检测沙门菌核酸等。另外,对于培养阳性的菌株和荧光PCR阳性的样品培养液进行了纳米孔宏基因组测序。
(一)通过培养、荧光PCR及长片段宏基因组测序,鉴定到发热病人中的伤寒和非伤寒沙门菌感染、以及混合感染。
加强监测期间,共收集272份发热病人血液样品,其中2份血培养液呈现荧光PCR阳性,从其中的1份分离培养到沙门菌。对荧光PCR阳性但培养未获得沙门菌的样品,利用纳米孔测序仪进行了宏基因组测序,获得3.2G数据,从中比对到了恶臭假单胞菌、大肠杆菌及沙门菌的唯一序列,进一步分析确定其中沙门菌的序列中含有O:54抗原合成基因簇序列,表明该样品为混合感染病例,但其所感染沙门菌不是伤寒和副伤寒沙门菌、而是非伤寒沙门菌。选取公共数据库中恶臭假单胞菌、大肠杆菌及沙门菌的参考序列,对样品中的序列进行了覆盖度的分析(图1),同时,利用宏基因组序列,也预测了样品中耐药基因的存在情况。
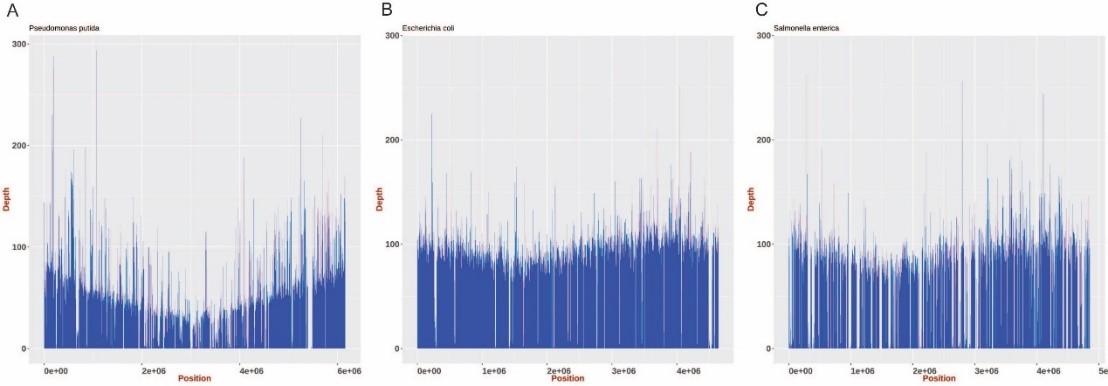
图1 样品中恶臭假单胞菌、大肠杆菌及沙门菌的序列覆盖度
(二)利用纳米孔长片段测序获得了伤寒沙门菌的全基因组序列和耐药基因变异
对分离到的沙门菌菌株,利用纳米孔测序仪进行了单菌测序,获得2G数据量,其中最长读长达到190.7Kb。经过拼接,获得了4.8Mb完成图,基于序列的血清型预测其为伤寒血清型,发现多个耐药基因(图2),推测可能耐氨基糖苷类、苯丙醇、甲氧咔胺嘧啶和磺胺类抗生素,并发现其还携带parC p: E84K 和gyrA p: A67P染色体位点突变,会造成对喹诺酮耐药。比对发现至少17个前噬菌体区域。将此株菌序列和105株来自全球的伤寒沙门菌序列构建系统发生树,可以看到该株菌和分离自西非尼日利亚、贝宁、布基纳法索和利比里亚的菌株聚为一簇,强烈提示该克隆群可能在西非已经传播流行数年(图3)。
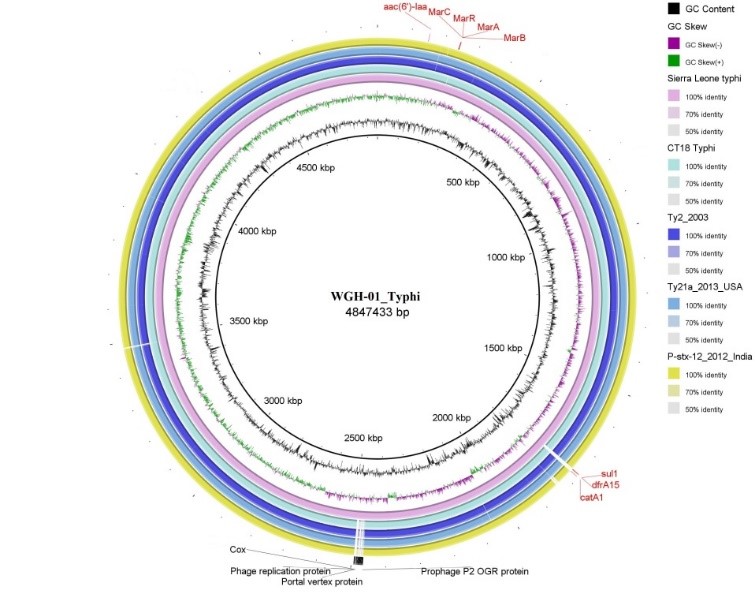
图2 塞拉利昂伤寒分离株的耐药基因分布情况及与公共数据库中其它伤寒沙门菌完成图的比较结果
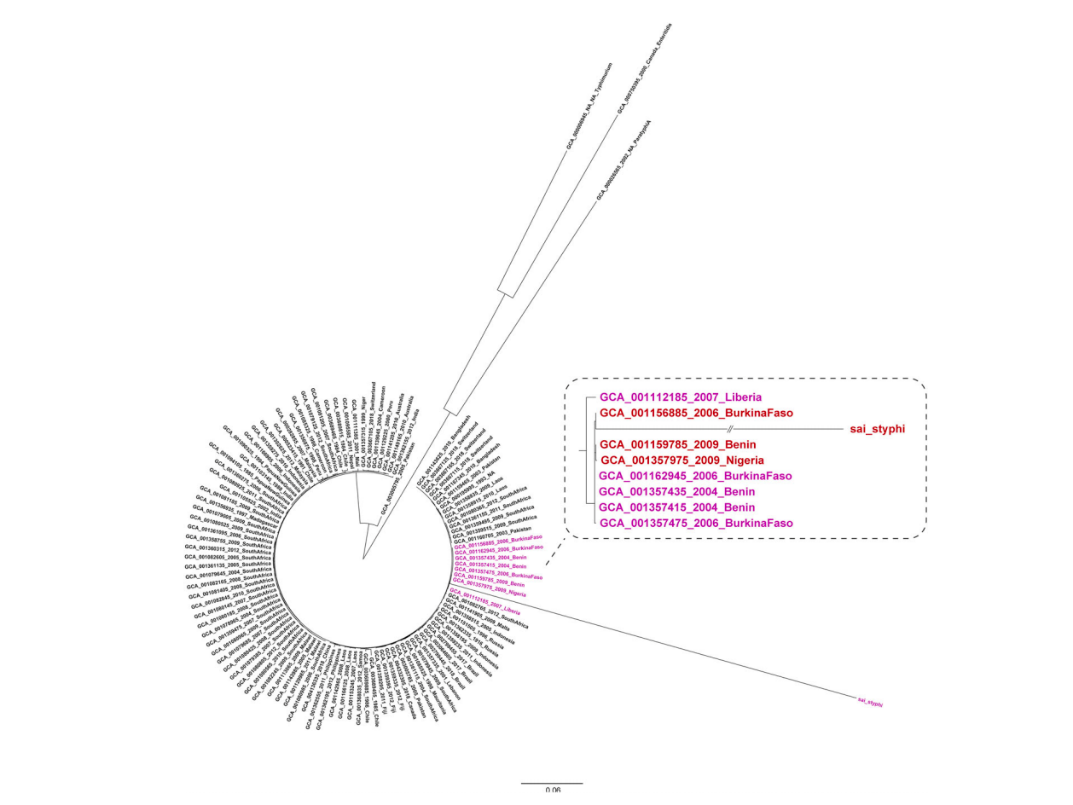
图3 塞拉利昂伤寒分离株与105株全球代表性伤寒沙门菌株进化树
(三)肥达试验导致了很高的假阳性判断
同时,在项目所收集的272份病人样品中,哨点医院对87份病人样品进行了肥达反应,其中78份显示肥达阳性结果,但这78份血液样品在荧光PCR和培养中均为阴性,显示肥达实验结果可信度很低,尤其在低流行人群中,可导致明显虚高的感染率判断。实际上,肥达试验可能和疟疾感染之间存在交叉反应,而塞拉利昂属于疟疾高发区,采用肥达反应诊断发热病例、会更容易造成误判。
(四)应在非洲地区建立必要的感染诊断参比实验室能力
通过本研究,获得了以下在伤寒感染诊断、流行确认以及公共卫生实验室能力建设方面的认识:
1. 在监测期间,弗里敦还没有发生伤寒的流行。但应注意分离到的菌株为西非流行多年的克隆,且很可能具有多耐药特征,仍存在感染和扩散风险。
2. 肥达反应操作简便、便于普及,但在伤寒低流行期、以及疟疾流行地区,应慎重使用,依靠肥达反应、在低流行期容易导致伤寒发病率的高估,需要加强操作质控、并采集双份血进行测试。
3. 纳米孔测序显示了很好的应用价值,通过宏基因组测序判断出了侵袭性沙门感染、提示可能存在混合感染、并获得了感染伤寒沙门菌菌株的基因组学与耐药基因特征。另外,基因组数据分析也解释了尽管在增菌样本中扩增检测到沙门菌核酸序列、但因其他菌的菌量高、未分离到沙门菌的原因。
4. 在非洲地区,也可以在不具备完善条件的实验室中、携带相应试剂开展纳米孔测序,进行感染病原诊断和发现,也可通过数据传输在高计算能力实验室完成分析。本研究中,是通过在北京的专业人员通过远程视频指导无测序经验的人员完成整个测序,并且将测定数据远程传回进行分析,清晰获得了感染病原信息。
该监测分析也提交到塞拉利昂卫生部,提出了伤寒诊断问题和建议,以及建立必要的病原分离、核酸检测参比实验室,甚至装备简便易行的测序系统,对感染病人开展病原学鉴定,能明确具体病因,提升治疗效果,避免抗生素滥用,并用于评估流行风险,便于政府及早制定相应的公共卫生措施。
以上研究以Molecular diagnostics and next-generation sequencing reveal real etiological characteristics of invasive Salmonella infection in febrile illness in Freetown, Sierra Leone为题发表在12月出版的Emerging Microbes & Infections杂志上(2022年 IF=19.6),河南疾病预防控制中心赵嘉咏、中国疾控中心传染病预防控制所卢昕、中塞友好固定生物安全实验室Alie Tie和Esther Ngegba,以及中国疾控中心王立立为共同第一作者,中国疾控中心董小平、萨拉里昂卫生部Doris Harding、中国疾控中心传染病预防控制所阚飙为共同责任作者。





