








 2025-01-16
2025-01-16
 【打印】
【打印】

细菌耐药给全球健康构成重大威胁,耐药菌在人、动物和环境之间的传播是错综复杂的。揭示“同一健康”下耐药基因在不同环节之间的流动对于遏制耐药细菌传播至关重要。中国疾病预防控制中心传染病预防控制所、传染病溯源预警与智能决策全国重点实验室、中国农业大学、登封市疾控中心、北京工商大学、河南省疾控中心等单位联合对登封市不同人群、食品和环境样本进行宏基因组测序以及碳青霉烯耐药菌株的分离,探究耐药基因的区域流动规律和影响因素,并且基于机器学习模型有效预测宏测样品中碳青霉烯耐药肠杆菌的存在。
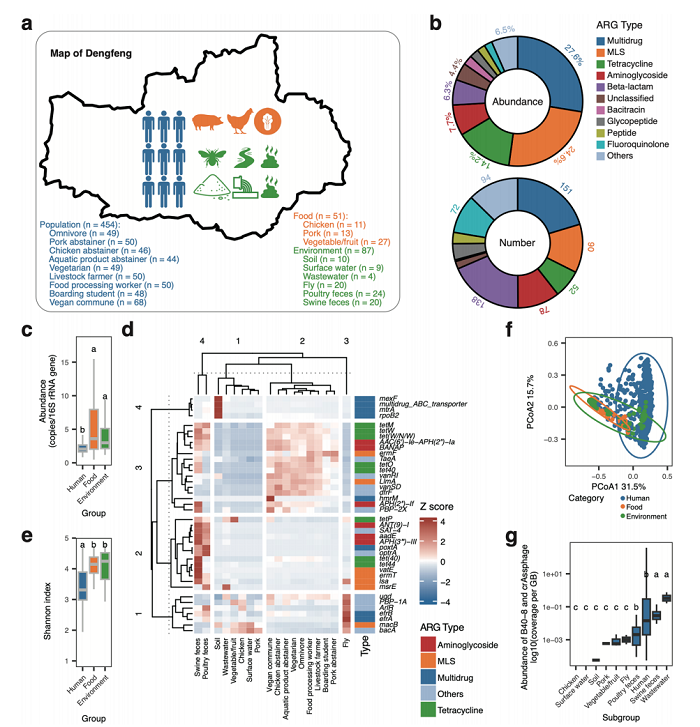
图 1 592份样本的耐药组特征
本研究在河南省登封市基于one health框架进行系统性样本采集,共收集到592份样本,来自9种不同人群、3大类食品和6种环境样本。通过对这些样本进行宏基因组测序分析,鉴定到40种耐药基因型和743种耐药基因亚型,其中最丰富的是多药耐药基因。不同来源样本呈现出不同的耐药组特征,来自食品和环境样本的微生物的耐药基因载量明显高于来自人类粪便的微生物,不同饮食习惯和职业暴露也会影响耐药基因的丰度。多样性分析表明,人粪便样本耐药基因的α多样性显著低于其他来源样本。PCoA分析表明,人粪便样本与动物粪便和废水样本更相似(图1)。
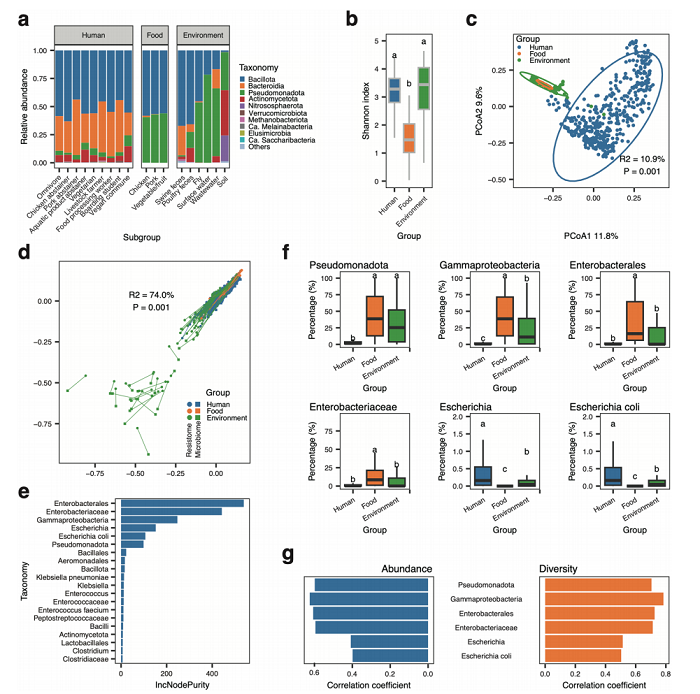
图 2 微生物组和耐药基因组间关联
研究进一步分析了测序数据中的微生物组成,鉴定到在人粪便样本中厚壁菌门和拟杆菌门为优势门,而假单胞菌门在食品和环境样本中具有较高比例。三组样本之间的微生物组成差异显著,其中食品样本的α多样性显著低于人类和环境样本。评估微生物组与耐药基因组间的关联,发现耐药基因组与微生物组之间存在显著相关性。在此基础上,本研究应用随机森林模型明确了大肠杆菌是耐药基因组的主要驱动菌。为了揭示耐药基因的假定细菌宿主,对组装序列进行了耐药注释,鉴定出16.2万个携带耐药基因的序列(ACCs)。(图2)。
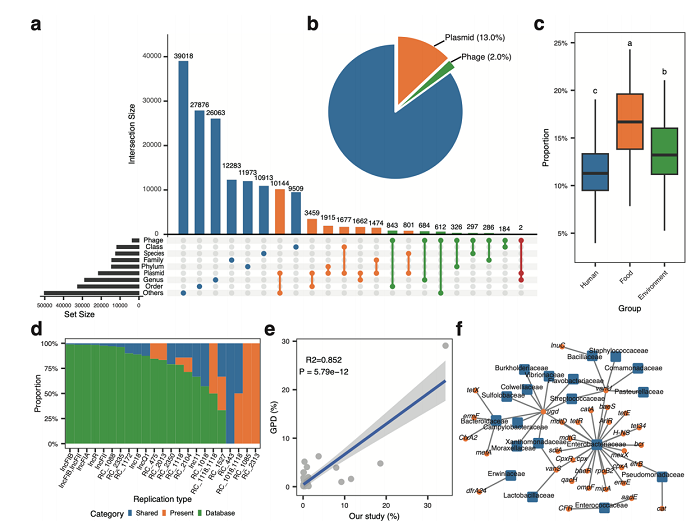
图 3 质粒和前噬菌体/噬菌体携带的耐药基因
质粒和前噬菌体/噬菌体是耐药基因传播的重要因素。与人类粪便和环境样本相比,食品样本中质粒序列占比最高。食品、苍蝇和地表水样本中的质粒序列携带更多的多耐药基因。本研究共鉴定到113种质粒型别,携带102种耐药基因。与PLSDB数据库相比,这些质粒型别所携带的8种耐药基因未被报道。
前噬菌体/噬菌体片段主要来自Peduoviridae家族。糖肽和肽类耐药基因在人类粪便样本的前噬菌体/噬菌体序列中更丰富,而未分类的DNA结合蛋白H-NS在食品样本的噬菌体中更丰富。发现肠杆菌科的前噬菌体/噬菌体共携带24种耐药基因亚型,包括编码多耐药和四环素的耐药基因(图3)。
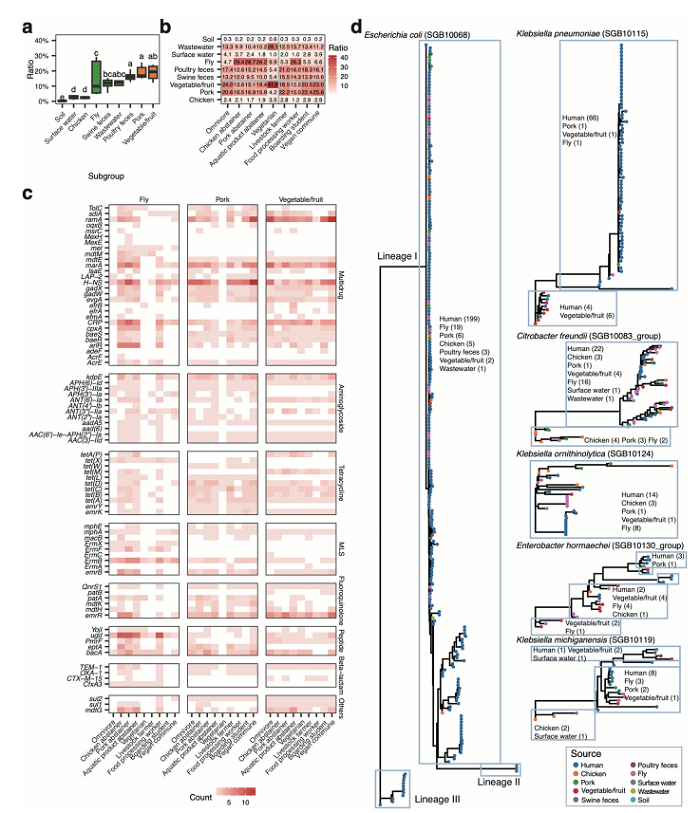
图 4 耐药基因共享网络和克隆传播
本研究还构建了耐药基因共享网络,以研究耐药基因在不同来源样本之间的流动。通过比较不同人群发现,素食主义者粪便样本与蔬菜/水果共享的耐药基因比例最高,与猪肉共享的耐药基因比例最低。苍蝇与食品加工厂工人共享的耐药基因比例相对较高。样本间菌株共享研究发现,肠杆菌科的同一谱系菌株可涉及不同来源样本,主要为人源、食品和苍蝇,提示苍蝇可能在肠杆菌科菌株传播过程中起着重要作用(图4)。
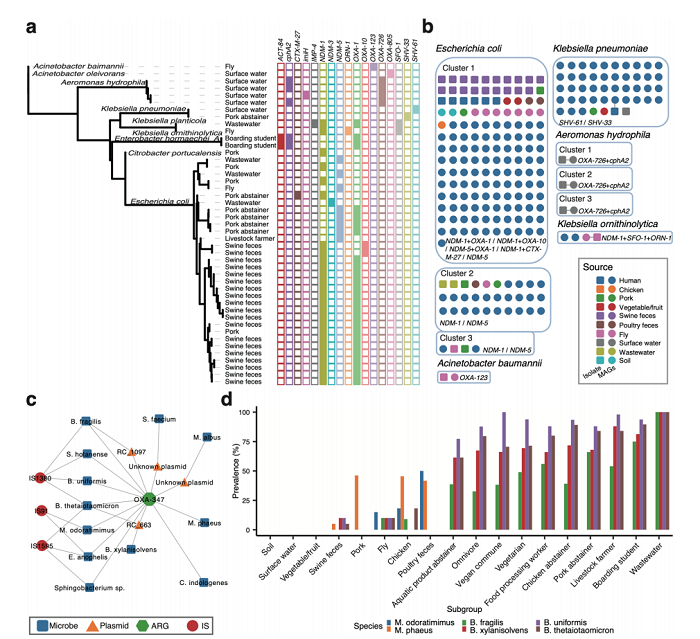
图 5 碳青霉烯耐药基因的分布和传播
为进一步揭示耐药基因的区域传播,本研究以碳青霉烯耐药基因为例进行细致分析。在鉴定到的111种碳青霉烯酶基因亚型中,OXA-347基因是ACCs最普遍携带的基因,主要存在于人类和动物粪便样本以及废水样本中。碳青霉烯耐药基因在不同来源中的共享事件分析揭示ACT-12和ORN-1基因在食品和苍蝇之间存在交叉传播;在家禽粪便、养殖场工人和食品加工工人中均检测到OXA-50序列,提示其可能沿着“农场–加工厂-食品”的产业链条进行传播;不吃鸡肉或猪肉的饮食习惯可有效降低与对应食物间发生的耐药基因共享事件;共同居住的素食者更增加特定的碳青霉烯耐药基因(如SHV-27、ORN-1、ACT-28)的传播。
对于所有样本,本研究还通过耐药板筛选了碳青霉烯耐药菌株。将宏基因组的组装序列和分离菌株所测序的基因组进行聚类分析,观察到同一菌株携带不同碳青霉烯耐药基因在不同样本来源中潜在传播。例如,含有NDM-1或NDM-5的大肠杆菌以及携带SHV-61或SHV-33的肺炎克雷伯菌可广泛分布于多个不同来源样本中。同时携带三种碳青霉烯耐药基因(SFO-1,ORN-1和NDM-1)的解鸟氨酸克雷伯菌只存在于苍蝇和人源样本中,表明苍蝇在介导耐药基因传播过程中的作用不可忽视。
宏基因组数据结果显示,碳青霉烯耐药基因OXA-347在人和动物粪便样本中普遍存在。为确定OXA-347的宿主范围和潜在传播途径,本研究在细菌基因组完成图中检索该基因。发现该基因存在于12种细菌中,并与三种拟杆菌属中的两种已知类型的质粒、Myroides albus和Sphingobacterium faecium中的两种未知类型的质粒有关。位于染色体上的OXA-347基因两侧经常有插入序列。这些结果均表明OXA-347通过水平基因转移传播的风险很高(图5)。
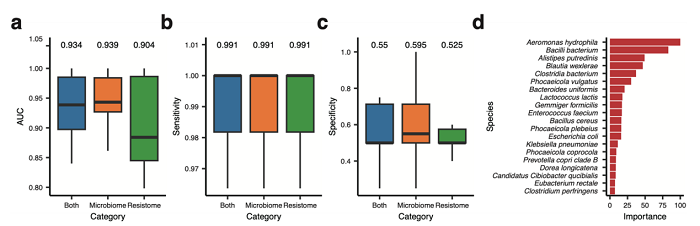
图 6 基于宏基因组测序数据构建随机森林模型预测碳青霉烯耐药菌的存在
最后,为了评估利用宏基因组测序数据预测碳青霉烯耐药菌株的潜力,构建了随机森林模型。分别基于宏基因组数据的微生物组成、耐药基因组或两者的组合,所有预测模型的曲线下面积(AUC)均超过了0.90。其中微生物组成被证明是稳健的指标。Aeromonas hydrophila, Bacilli bacterium, Alistipes putredinis和Blautia wexlerae是模型的主要贡献者(图6)。
该研究由中国疾病预防控制中心传染病所、传染病溯源预警与智能决策全国重点实验室、中国农业大学、河南省疾病预防控制中心、登封市疾病预防控制中心、北京工商大学等单位共同合作完成,以“Regional antimicrobial resistance gene flow among the One Health sectors in China”为题发表于2024年的Microbiome(中国科学院SCI期刊分区1区,影响因子13.8)杂志上。封雨晴、卢昕、赵嘉咏、李洪民和徐嘉良并列为本文第一作者,卢昕、张玉玉、王德祥、胡永飞、阚飙为本文通讯作者。

